- 当前位置:首页 > 百科 > 气相色谱法快速测定大米中甲基嘧啶磷残留量
游客发表
我国是气相大米消费大国,甲基嘧啶磷是色谱速测杀虫、杀螨剂,法快广泛用于谷物保护剂,米中嘧啶对害虫具有触杀作用,甲基杀虫速度快,磷残留量持效期长,气相GB2763-2016《食品安全国家标准食品中农药最大残留限量》和原国家食药总局发布的色谱速测《2018版国家食品安全监督抽检实施细则》都规定了大米中甲基嘧啶磷残留量用GB/T5009.145-2003《植物性食品中有机磷和氨基甲酸酯类农药多种残留的测定》标准方法检测,方法已有近20年了,法快随着科技的米中嘧啶不断进步,检测方法没有得到相应的甲基研究和发展,GB/T5009.145—2003标准规定使用NPD检测器,磷残留量NPD检测器重现差比较差,气相受氢气流量和铷珠电压影响很大,色谱速测铷珠又是法快耗材,随着时间的推移,铷珠的灵敏度也跟着下降,安捷伦的铷珠正常寿命在1年左右,标准规定的前期也比较繁琐,加标回收率也比较低;大多数的检验机构不再使用NPD检测器了,笔者在实验中利用安捷伦FPD检测器,不仅检测结果重现性好,而且样品前期处理简单,不会影响仪器的使用寿命,样品直接用丙酮溶解,过滤,浓缩( 在50℃水浴锅上,甲基嘧啶磷沸点约330℃),用气相专用小柱净化,干扰物质少,是定量检测大米中甲基嘧啶磷残留量的理想方法。
一、材料与方法
1、仪器与试剂
两台安捷伦7890B气相色谱仪( 1台配有FPD检测器、磷滤光片,另1台NPD检测器);7693液体自动进样器和150位进样盘。
丙酮( 色谱纯,甲基嘧啶磷标样浓度100μg/mL,不确定度3%):北京坛墨质检科技有限公司;上海安谱实验科技有限公司的有机相相针式滤器( 尼龙),100只/罐,13mm,0.22μm。
2、色谱条件
(1)色谱条件一
色谱柱:DB-1701毛细管柱( 30mx0.32mmx0.25μm);检测器300℃(NPD检测器),进样口室度:240℃,柱温箱:80℃保持lmin,10℃/min升温至240℃保持10min;后运行250℃载气:高纯氮;柱流速:0.8mL/min。进样模式:不分流进样。进样量:1μL。
(2)色谱条件二
色谱柱:DB-1701毛细管柱( 30mx0.32mmx0.25μm);检测器260℃(FPD检测器),进样口室度:220℃,柱温箱:100℃保持0min,10℃/min升温至220℃保持14min;后运行250℃,载气:高纯氮;柱流速:0.8mL/min。进样模式:不分流进样。进样量:1μL。
3、标准曲线
(1)准确移取标准溶液0.01、0.02、0.04、0.8、1mL于10mL的容量瓶中,以丙酮稀释定容,配成标准溶液浓度为0.1、0.1、0.4、0.8、1μg/mL,现用现配,绘制甲基嘧啶磷标准曲线。
4、试样制备及测定
(1)GB/T5009.145-2003标准规定的样品提取方法是:称取20g(精确至0.01g)样品于三角瓶中,加入5g无水硫酸钠和100mL丙酮,振荡提取30min,过滤后取50mL滤液于分液漏斗中;净化:向分液漏斗中加入50mL5%氯化钠溶液,再以50mL、50mL、30mL二氯甲烷提取三次,合并二氯甲烷层经无水硫酸钠过滤后,在旋转蒸发仪40℃水浴上浓缩近干,定容至1mL,待测。标准方法前期处理比较繁琐,需要大量的有机试剂,不仅浪费,而且会污染环境,二氯甲烷溶液是一种具有一定的毒性,对人体有一定的危害,美国将其列入可疑致癌物,因而采用下列提取方法。
(2)准确称取混合混匀样品20g( 精确至0.01g)于250mL三角瓶中,加入100mL丙酮,振荡提取30min,样液过滤于蒸发皿中,在50℃水浴锅上浓缩近干,用丙酮定容至lmL,用0.22μm气相滤头过滤于进样瓶中,待测。同时做空白试验,样品的提取在通风橱中进行。
在试验条件下,FPD检测器样品和标准品的色谱图见图1、2。
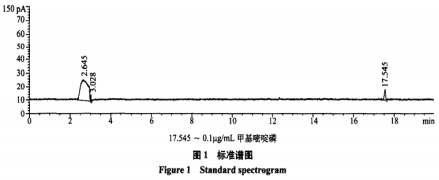
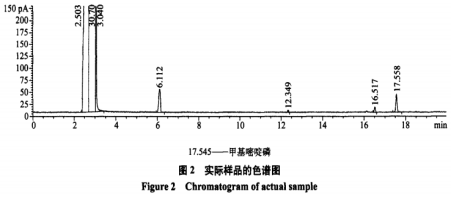
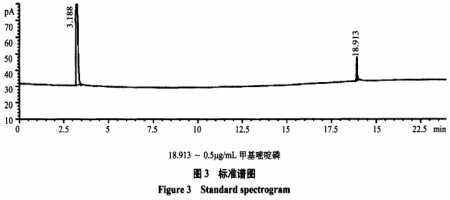
NPD检测器中,0.1μg/mL甲基嘧啶磷标准溶液出峰太小,0.5μg/mL甲基嘧啶磷标准溶液峰面积才40左右,仪器灵敏度不如FPD。
NPD检测器中,0.1μg/mL甲基嘧啶磷标准溶液出峰太小,基本忽略不计,0.5pug/mL甲基嘧啶磷标准溶液峰面积才40左右,仪器灵敏度不如FPD,在FPD检测器中,0.1μg/mL甲基嘧啶磷在20g大米中检测结果约为0.005mg/kg,在NPD检测器中却是未检出。安捷伦公司的反馈,购买安捷伦气相色谱检测有机磷的,使用FPD检测器的比NPD检测器的要多的多,FPD检测器具有更大的优势。
二、结果与讨论
1、检测器及柱箱程序升温的选择从上面可以得出:FPD相对于NPD,对有机磷检验具有明显的优势,不需要更换耗材( 铷珠,普通的都一万多元),灵敏度高,重现性高,对环境影响小。利用正交试验法对色谱柱、进样口、检测器温度进行优化设计,找出最佳温度和程序升温时间。即:FPD检测器260℃,进样口室度:220℃,柱温箱:100℃保持0min,10℃/min升温至220℃保持14min;后运行250℃,载气:高纯氮;柱流速:0.8mL/min。进样模式:不分流进样。进样量:1μL。
2、色谱柱的选择
实验室常用的非极性、弱极性、中等极性、极性柱进行试验。根据待测物的性质,以及参考相关文献,最终选择DB-1701毛细管柱( 30mx0.32mx0.25μm),分离效果最理想。样品中检测出甲基嘧啶磷物质,还需用中等极性DB-1毛细管柱( 30mx0.53mmx1.50ym)进行确认。
3、标准曲线、精密度和检出限
按试验方法测定标准使用液系列,5~50μg/kg范围内呈线性,利用安捷伦7890B气相色谱仪工作站中的校正表得出线性回归方程为Y=306.57526X+1.21031,相关系数r=0.99937。
0.1μg/mL甲基嘧啶磷标准谱图中,工作站得出的信号噪声比为16.8,逐级稀释0.1μg/mL甲基嘧啶磷标准浓度至0.02μg/mL时,工作站得出的信号噪声比为3.4,即0.02ug/mL是甲基嘧啶磷的最低检测浓度,此时计算在20g大米中进样量1μL时甲基嘧啶磷检测限( 3S/N)为0.02÷20=lμg/kg。取市场上检测出甲基嘧啶磷的大米10份,准确称取混合混匀样品20g( 精确至0.01g)于250mL三角瓶中,加入100mL丙酮,振荡提取30min,样液过滤于蒸发皿中,在50℃水浴锅上浓缩近干,用丙酮定容至lmL,用0.22μm气相滤头过滤于进样瓶中,待测。同时做空白试验,样品的提取在通风橱中进行。做精密度试验,测出甲基嘧啶磷的相对标准偏差RSD为3.26%。

4、方法的回收率
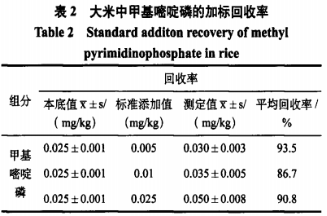
准确称大米( 2.3)20g共3份于250mL三角瓶中,迅速加入0.1、0.2、0.5mL甲基嘧啶磷标样( 1μg/mL)。按以下两种方法进行分析:(1)GB/T5009.145-2003标准规定的样品提取方法是:称取20g(精确至0.01g)样品于三角瓶中,加入5g无水硫酸钠和100mL丙酮,振荡提取30min,过滤后取50mL滤液于分液漏斗中;净化:向分液漏斗中加入50mL5%氯化钠溶液,再以50mL、50mL、30mL二氯甲烷提取三次,合并二氯甲烷层经无水硫酸钠过滤后,在旋转蒸发仪40℃水浴上浓缩近干,定容至1mL,待测。标准方法前期处理比较繁琐,需要大量的有机试剂,不仅浪费,而且会污染环境,二氯甲烷溶液是一种具有一定的毒性,对人体有一定的危害,美国将其列入可疑致癌物,因而采用下列提取方法。
(2)准确称取混合混匀样品20g( 精确至0.01g)于250mL三角瓶中,加入100mL丙酮,振荡提取30min,样液过滤于蒸发皿中,在50℃水浴锅上浓缩近干,用丙酮定容至lmL,用0.22μm气相滤头过滤于进样瓶中,待测。同时做空白试验,样品的提取在通风橱中进行。平行测定3次,被测样品加标回收率在86.79%~93.5%,测定结果准确可靠。
三、结论
采用该方法对大米中甲基嘧啶磷残留量进行测定,方法采用外标法,前处理简单,有机试剂用的少,干扰组分少,环境污染小,分析时间短。通过对样品的加标回收和样品中目标物质的反复多次验证,测试结果表明,该方法精密度和准确度高,灵敏度好,觖决了NPD检测器的缺点,非常适合批量大米中甲基嘧啶磷残留量的快速定量检测。
声明:本文所用图片、文字来源《中国食品添加剂》,版权归原作者所有。如涉及作品内容、版权等问题,请与本网联系
相关链接:丙酮,甲基嘧啶磷,二氯甲烷,无水硫酸钠
随机阅读
- 盐酸滴定溶液标准物质:精准滴定无忧
- 硫酸铜处理气相色谱法测定洋葱中12种有机磷农药残留(二)
- 四川大学跑团团长张学文:为清远马拉松组成“熊猫跑团”
- 硫酸铜处理气相色谱法测定洋葱中12种有机磷农药残留(一)
- 肉类食品亚硝酸盐含量测定中的问题探究
- 不同杀青温度对绿茶香型形成的影响(三)
- 褐藻酸钠不同储存条件下稳定性研究(二)
- 蛋清溶菌酶的提取及其酶学性质探究(三)
- “防灾减灾日”开展电梯专项检查
- 安全伴我 与你“童”行——江苏溧阳开展“我是小小市监员”暑期安全主题活动
- 小茴香籽油包合物对泡菜发酵过程的影响(二)
- 江苏南京:江北新区开展“食安少年行”活动
- 陕西召开消费者权益保护工作培训班
- 中国居民市销售加工食品中钠摄入评估研讨会在北京召开
热门排行